
FDA 计划发布 CAR-T 治疗产品制造指南
美国 FDA 生物制品审评与研究中心(CBER)主任 Peter Marks 表示,FDA 正在制定关于 CAR-T 细胞疗法生产的新指南以提供更清晰的监管期望信息。但他表示尚不确定指南什么时候会发布出来。
日前,在遗传工程和生物技术新闻(GEN)举办的题为“生物制造在细胞和生物治疗产品中起核心作用”的网络研讨会上,Marks 分享了 CBER 计划如何在生物产品的关键开发和制造阶段以及整个产品生命周期中监管这些产品。
Marks 首先介绍了美国基因治疗产品的市场现状,目前共有五个基因治疗产品获批,其中三个为 CAR-T 产品。详细分析请见【识林案例解析:已上市CAR-T产品】。我国 CAR-T 产品研发现状详见【识林案例解析:CAR-T国内研发现状】。更多 CAR-T 法规指南、案例解析和资讯等内容请登录识林搜索“CAR-T”主题词查看。
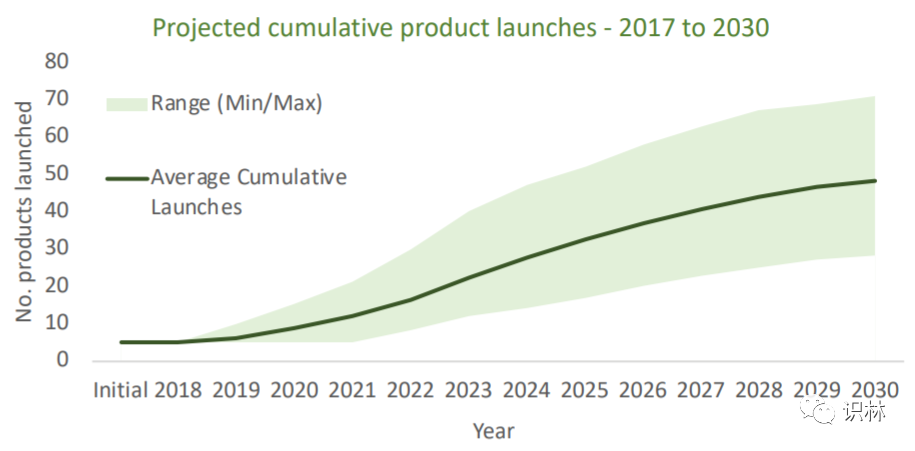
基因治疗产品领域迅猛发展,截至 2020 年 7 月,有超过 1000 件活跃 IND。尽管新冠疫情肆虐,但并没有影响到基因治疗产品 IND 产品的提交热情,仅 2020 年上半年就有 134 件 IND 提交到 FDA,照此估算,2020 年的全年提交量将会超过 2019 年(245 件)。根据 MIT Newdigs FoCUS 预测,到 2030 年将有 40 到 60 个商业化基因治疗产品上市(见下图)。
关于 FDA 如何监管在护理点生产的细胞和基因疗法的话题,Marks 表示,FDA 有可能监管制造治疗产品的设备,而不是对每个拥有该设备的场所都要求生物制品许可申请(BLA)。Marks 表示,“存在的问题是,设备将使你能够在护理点制造产品,而许可或批准该设备可能会解决这一问题。另一方面,更复杂的问题是,是否可以在护理点制造 CAR-T 细胞。这是一个更具挑战性的问题。”
Marks 解释指出,将有两种可能的监管模式,一是许可单个护理点;二是有一个持有许可的主要申办人,而护理点设施成为该许可下的“制造子场地”。Marks 建议,无论哪种方式,如果企业考虑走其中一条路,那么最好与 FDA 就可能存在关键问题进行讨论。
Marks 认为,FDA 目前发现的与细胞和基因疗法制造相关的最常见的问题是,企业如何通过不同的产品开发阶段从一代制造过程发展到下一代。“我们对制造流程的发展持非常开放的态度,但是问题是,有时候,拥有第一代制造流程的人会获得非常好的临床数据,然后他们不得不采用新的流程来扩大规模,规模放大会带来问题,而由于已经进行了重大流程变更,以至于在需要进行桥接试验时无法桥接。”基因治疗产品的制造问题 Marks 与相关企业负责人已经讨论过多次:【制造环节:细胞与基因疗法面临的下一个大挑战 2019/01/24】,【基因治疗产品开发瓶颈:缺乏明确灵活的监管框架 2020/06/23】。
Marks 对于考虑这些问题的制造商的建议是,“在可能的范围内,在进行流程开发时,尽可能地未雨绸缪,并尝试将你希望使用的流程尽快迁移到用于商业产品的生产。”另外,Marks 鼓励细胞和基因治疗产品的开发者对于转向更大规模的商业化制造流程制定合理的经过验证的计划。
关于效价测定问题,Marks 强调了在 3 期试验之前使用经过验证的测定的重要性。他表示,“我们非常希望看到在 3 期试验时能够有经过验证的效价测定。这在后期非常有帮助。有时候,有些开发者最终无法验证其效价测定,使得这最终成为一个问题。”
对于那些希望将新技术(例如 , 下一代测序技术和用于病毒控制的高通量系统)纳入其开发计划的企业,Marks 鼓励企业提请召开 CBER 先进技术团队会议,“我们非常愿意举行关于这些平台的会议”而且这些会议“非常有用”。他表示 , “由于会议是非正式的,我们发现申办人往往会收获很多 , 而且实际上我们往往收获更多,因为我们了解了申办人的新技术。”
作者:识林-椒
