
FDA plans to publish CAR-T therapeutic product manufacturing guidelines
Peter Marks, director of the US FDA Center for Biologics Evaluation and Research (CBER), said that the FDA is developing new guidelines for the production of CAR-T cell therapy to provide clearer regulatory expectations. But he said it is not yet certain when the guidelines will be released.
A few days ago, in a webinar titled "Biomanufacturing plays a central role in cells and biotherapeutic products" organized by Genetic Engineering and Biotechnology News (GEN), Marks shared how CBER plans to develop and manufacture key biological products. Supervise these products in stages and throughout the product life cycle.
Marks first introduced the current market status of gene therapy products in the United States. There are currently five gene therapy products approved, three of which are CAR-T products. For detailed analysis, please see [Case Analysis of Shilin: CAR-T Products Listed]. For details of the current status of CAR-T product research and development in my country, please refer to [Silin Case Analysis: CAR-T Domestic Research and Development Status]. For more CAR-T regulations, guides, case analysis and information, please log in and search for "CAR-T" subject headings to view.
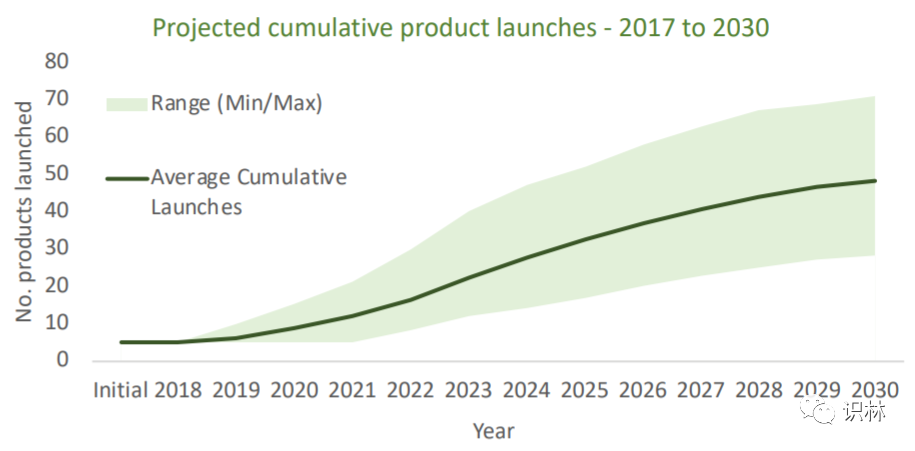
The field of gene therapy products is developing rapidly. As of July 2020, there are more than 1,000 active INDs. Despite the raging new crown epidemic, it did not affect the enthusiasm for gene therapy product IND submissions. In the first half of 2020 alone, 134 INDs were submitted to the FDA. Based on this estimate, the number of submissions in 2020 will exceed 2019 ( 245 pieces). According to the prediction of MIT Newdigs FoCUS, there will be 40 to 60 commercial gene therapy products on the market by 2030 (see the figure below).
Regarding the topic of how the FDA regulates cell and gene therapies produced at the point of care, Marks stated that it is possible that the FDA may regulate equipment that manufactures therapeutic products, instead of requiring a biological product license application (BLA) for every location that has the equipment. Marks said, “The problem is that the device will enable you to manufacture products at the point of care, and licensing or approval of the device may solve this problem. On the other hand, the more complicated question is whether CAR can be manufactured at the point of care. -T cells. This is a more challenging problem."
Marks explained that there will be two possible regulatory models, one is to license a single point of care; the other is to have a main sponsor with a license, and the point of care facility becomes a "manufacturing sub-site" under the license. Marks suggested that, either way, if a company considers one of these paths, it is best to discuss with the FDA about possible key issues.
Marks believes that the most common problem found by the FDA related to cell and gene therapy manufacturing is how companies can evolve from one-generation manufacturing process to next-generation through different product development stages. "We are very open to the development of the manufacturing process, but the problem is that sometimes, people with the first generation of manufacturing process will get very good clinical data, and then they have to adopt a new process to scale up and scale up. It will cause problems, and because major process changes have been made, it is impossible to bridge when bridging tests are required." Marks has discussed the manufacturing issues of gene therapy products with relevant business leaders many times: [Manufacturing link: cell and The next big challenge for gene therapy 2019/01/24], [Gene therapy product development bottleneck: lack of clear and flexible regulatory framework 2020/06/23].
Marks’s advice to manufacturers considering these issues is, “As far as possible, when developing processes, try to be as proactive as possible, and try to migrate the processes you want to use to the production of commercial products as soon as possible.” , Marks encourages cell and gene therapy product developers to formulate reasonable and proven plans for shifting to larger-scale commercial manufacturing processes.
Regarding the issue of potency determination, Marks emphasized the importance of using validated assays before phase 3 trials. He said, “We really want to see a validated potency measurement in the phase 3 trial. This is very helpful in the later stage. Sometimes, some developers cannot verify their potency measurement in the end, making this ultimately a problem ."
For those companies that want to incorporate new technologies (for example, next-generation sequencing technologies and high-throughput systems for virus control) into their development plans, Marks encourages companies to invite CBER advanced technology team meetings, "We are very willing to hold meetings on these platforms Meetings" and these meetings are "very useful". He said, "Because the meeting is informal, we find that sponsors tend to gain a lot, and in fact we tend to gain more because we understand the sponsor's new technology."
Author: Lin know - pepper
